Epigenetic Analysis
BEA offers solutions for studies of gene regulation on the epigenetic level with the most commonly used methods for epigenetic studies.
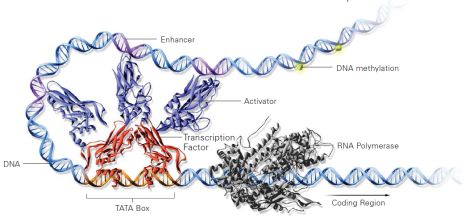
ChIP-Sequencing
By combining chromatin immunoprecipitation (ChIP) assays with sequencing, ChIP sequencing (ChIP-Seq) is a powerful method for identifying genome-wide DNA binding sites for transcription factors and other proteins. ChIP-Seq enables thorough examination of the interactions between proteins and nucleic acids on a genome-wide scale. Sequence analysis of bound DNA targets for transcription factors or histone modifications across the entire genome of any organism can reveal gene regulatory networks in combination with RNA sequencing and methylation analysis.
We construct ChIP sequencing libraries using the NEBNext ChIP-Seq library reagent set for Illumina for standard input ( 0,25-5 ng DNA). For higher eukaryotes a minimum of 10-20 million mapped reads per sample are recommended for performing a whole genome ChIP analysis.
Reduced representation bisulfite sequencing (RRBS)
RRBS reduce the scale and cost of whole genome bisulfite sequencing by only sequencing a reduced, representative sample of the whole genome. DNA is first digested with a restriction enzyme such as MspI which is methylation sensitive and cuts at CCGG sites. The CpG-rich regions of the genome is thereby enriched. The DNA ends are then filled in and adapters are ligated. The DNA-fragments are then size selected, bisulfite converted, and sequenced. This simple approach captures 85% of CpG islands and 60% of promoters and requires very little input sample. The reduction in DNA also means that fewer reads are needed for accurate sequencing, reducing cost drastically. One disadvantage of RRBS is that the assay does not capture all CpG islands or promoters. RRBS has become popular when high-throughput, low cost methylation analysis is needed such as in clinical applications. Currently BEA is using Diagenode's Premium RRBS kit .

- Customer Information:
- Project Submission
- External Users
- iLabs
- NGS Sample Submission
- How to acknowledge BEA
- Reference List
- Latest Newsletter
- FAQs
- BEA Price List:
- Order Forms:
- Links:
- KI core facilities for research
- SICOF-Single cell core facility
- CBB-Centre for Bioinformatics and Biostatistics
- SciLifeLab
- National Bioinformatics Institute Sweden
| BEA Service Fees |  |
| BEA Services | |  |
